Background on FSHD
Facioscapulohumeral muscular dystrophy (FSHD) is a hereditary human myopathy, affecting groupes of muscles in the face and shoulder, mainly caused by the contraction of a macrosatellite array on chromosome 4q35. The mechanism by which this triggers the disease has long represented one of the most enigmatic conundrums for human geneticists. Progress has been made in recent years with the identification of DUX4, a transcription factor encoded in a repeated 4q35 macrosatellite, produced in excess in FSHD and toxic for muscle (comprehensive review in Tawil et al., 2014). DUX4 activation results from the combination of a polymorphism stabilizing its RNA, with additional modifications that enhance DUX4 transcription by interfering with a mechanism of epigenetic silencing. Patients with identical DUX4-activating contexts can vary a lot in disease severity, ranging from childhood onset with severe phenotypes, to asymptomatic elderly carriers. This implies the existence of additional genetic FSHD-modifier genes contributing disease onset and severity.
The 4q35 FAT1 gene is an FSHD-modifier gene
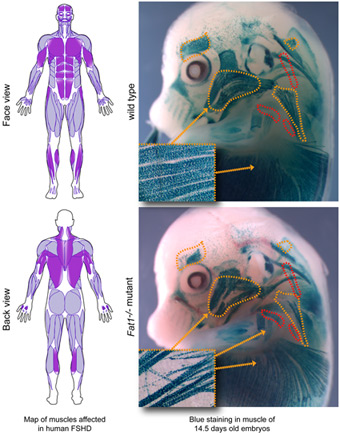
We initially identified the Fat1 Cadherin as a novel regulator of myogenesis in mice, controlling morphogenesis of subset of muscles (Caruso et al., Plos Genetics, 2013). The topology of muscle phenotypes exhibited by Fat1 mutant mice was highly reminiscent of the map of muscles affected in FSHD patients, and Fat1 mutant mice also exhibited non-muscular FSHD-like phenotypes (Caruso 2013). In collaboration with the groups of Nicolas Levy and Marc Bartoli (Marseille Medical Genetics Unit) and of Julie Dumonceaux (UCL), we collectively established a link between between dysregulation of FAT1 and FSHD: (1) we found that FSHD muscles exhibit lowered FAT1 levels at fetal (Caruso 2013) and adult (Mariot et al., Annals of Neurology, 2015) stages; (2) we identified pathogenic FAT1 variants in FSHD-like patients, affecting either RNA/protein structure (Puppo et al., Human Mutation, 2015), or deleting a putative cis-regulatory enhancer in the FAT1 gene (Caruso 2013). These data are compatible with the idea that FAT1 represents an FSHD modifier gene, which deregulation co-occurs with FSHD, thereby contributing to defining the topography of symptoms.
A more complete description of these findings is available here.
More on what Fat1 can teach us:
Following the first group of publications on Fat1, we (in fact, I, but this was always a collective intention, so I prefer saying we) continued exploring functions of Fat1 as a potential guide to learn more about the pathology. We first went deeper on its role during neuromuscular development, and took advantage of our conditional allele to dissect tissue-specificity of its functions. We first reexplored the muscle phenotype in the context of a specific muscle, that extends subcutaneously starting from the base of the forelimb (called CM, for cutaneous maximus). Fat1 was specifically expressed in the motor neuron pool innervating this muscle, making it the best cell type in which it might play a role, but also in the subcutaneous mesenchymal interface between muscle and skin within which this muscle migrated, in addition to myogenic cells, in which we had already found that Fat1 was acting to control migration polarity (Caruso et al, 2013).
Surprisingly, we found that activities in all three cell types contributed to the muscle phenotype (and associated motor neuron defects). In addition to our previous findings on myogenic cells, we then found that the strongest muscle phenotype occurred in embryos with mesenchyme-specifc deletion, severely impacting progenitor migration and myofiber extension, implying that a non-cell-autonomous activity in non-myogenic mesenchyme was key for muscle expansion. Motor-neuron-specific deletion also resulted in impaired growth of this muscle, while phenotypes in other muscles (not innervated by Fat1-expressing muscles) was not seen. Both of these activities (in mesenchyme and motor neurons) led to a reduction of Gdnf expression in the CM progenitors. Interestingly, Gdnf was required to induce Etv4 expression in these motor pools, thereby influencing their cell fate specification and axonal target innervation. As a result, Fat1 activities in both the mesenchyme (Fat1+ layer at muscle skin interface) and in CM motor neurons (Fat1+ too), they similarly impacted on the size of the corresponding motor neuron pool, identified by expression of the transcription factor Etv4, and within this pool, with the capacity of Etv4 to induce expression of one of its target genes, Clusterin. They also both impacted on innervation of the CM muscle, target of Etv4+ neurons.
So far, what was known was that specification of Etv4+ motor pools and their axonal growth toward and within their target muscle was induced by Gdnf, coming from the plexus mesenchyme and form the two target muscles of Etv4+ motor neurons (CM, and Latissimus dorsi, in which pool Fat1 was not expressed). Gdnf null mutants loose both Etv4 and Clusterin expression, and Etv4 null mutants lack Clusterin expression. The discovery that Fat1 controlled Gdnf expression in CM myogenic progenitors (through its activities in mesenchyme and motor neurons) suggested that GDNF might be a contributor of the motor neuron specification and innervation phenotypes. This idea was confirmed by the fact taht Gdnf heterozygocity (the Gdnf-lacZ allele used as a readout of Gdnf expression was a heterozygous knockout of Gdnf) enhanced the Clusterin expression phenotype of both MN-specific and mesenchyme-specific mutants. Thus, the reduction in expansion of the progenitor cells was a likely contributor (via size reduction of the source of Gdnf) of the cell fate phenotype of the CM motor pool. In adult mice, this also led to a reduction in size of the NMJs, another readout of GDNF activity, without effects of muscle fiber diameter.
Alltogether, this work showed that several distinct functions of Fat1 in different cell types contribute to components of a same global phenotype, reflecting the collaborative nature of cell types involved in assembly of neuromuscular circuits. This work was published by me in 2018 (Helmbacher, Plos Biology, 2018) and also covered in a blog post in the Node, where I gave a more historical account on these discoveries (link).
What about the Fat1 enhancer deleted in FSHD patients ?
Following the identification of an FSHD-associated Copy Number Varian (CNV) deleting a putative intragenic FAT1 enhancer, we have followed up this possibility and investigated the transcriptional activity of this putative enhancer in vivo. We found this enhancer to drive expression in developing myofibers and in muscle-associated mesenchymal cells at selective subset of muscle extremities (relevant to Fat1-deficiency phenotypes). We discuss the possible functional implications of such a deletion, and propose how heterozygous deletions of this enhancer may act as disease modifier when associated with FSHD (Caruso et al., 2022, biorxiv).
Fat1 in FAPs during muscle regeneration: a negative modulator of fibroadipogenesis
More recently, continuing the work in mouse to explore Fat1 functions, after having studied Fat1 contribution to developmental myogenesis, we next wondered if Fat1 was contributing to myogenic repair in adult skeletal muscle. This led us to uncover a new function for Fat1 in homeostasis of Fibro-adipogenic Progenitors (FAPs). FAPs are muscle-resident mesenchymal cells known both for their pro-myogenic activity in healthy muscle, and for contributing to fibro-fatty infiltrations in muscle pathologies. We found that Fat1 ablation in the mesenchymal lineage enhances fibro-adipogenic differentiation and adipose infiltrations after glycerol injury. The use of an inducible CRE line and lineage tracing showed that this negative control of adipogenesis involves both cell-autonomous and non-cell-autonomous functions. This identifies Fat1 as a novel modulator of fibro-adipogenic differentiation, and establishes mesenchyme-specific Fat1 mutants as a model of exacerbated fibro-fatty infiltrations after glycerol injury (Ferracci & Helmbacher, biorxiv, 2026).
Publications
2026
- Preprint: Pierre-Antoine Ferracci, Françoise Helmbacher. Fat1 deletion enhances Fibro-Adipogenic Differentiation and Adipogenic expansion following injury in skeletal muscle. biorxiv, (2026). April 2026 | DOI | PDF |
2022
- Nathalie Caruso, Angela K. Zimmermann, Tarana Nigam, Celine Becker, Karelia Lipson, Françoise Helmbacher. “An intragenic FAT1 regulatory element deleted in muscular dystrophy patients drives muscle and mesenchyme expression during development” Biorxiv (2022), September 17 | doi: 10.1101/2022.09.14.507898 | PDF |
2018
- Helmbacher F. Tissue-specific activities of the Fat1 cadherin cooperate to control neuromuscular morphogenesis. PLOS Biology (2018) 16(5) e2004734 | PMID: 29768404 | doi: 10.1371/journal.pbio.2004734 | preprint: https://doi.org/10.1101/207308 |
2018 (non-peer-reviewed)
- Blog post: Helmbacher F. Unraveling tissue interactions coordinating neuromuscular morphogenesis: a journey through serendipity Blog Post, The Node, 20th June, 2018
2015
- Puppo F., Dionnet E., Gaillard MC., Gaildrat P., Castro C., Vovan C., Bertaux K., Bernard R., Attarian S., Goto K., Nishino I., Hayashi Y., Magdinier F., Krahn, M., Helmbacher, F., Bartoli, M, and Levy, N. (2015). Identification of variants in the 4q35 gene FAT1 in patients with a Facioscapulohumeral dystrophy (FSHD)-like phenotype. Human Mutation, 2015, 23 JAN. DOI: 10.1002/humu.22760 | PMID 25615407 |
- Mariot V, Roche S, Hourdé C, Portilho D, Sacconi S, Puppo F, Rameau P, Caruso N, Delezoide AL, Desnuelle C, Bessières B, Collardeau S, Feasson L, Maisonobe T, Magdinier F, Helmbacher F., Mouly V, Butler-Browne G. Dumonceaux J. Correlation between low FAT1 expression and early affected muscle in FSHD. Annals of Neurology, 2015, MAY 28. | PMID: 26018399 |
2013
- Caruso N., Herberth B., Bartoli M., Puppo F., Dumonceaux J., Zimmermann A., Denadai S., Lebossé M., Roche S., Geng L., Magdinier F., Attarian S., Bernard R., Maina F., Levy N. and Helmbacher F. (2013). Deregulation of the protocadherin gene FAT1 alters muscle shapes: implications for the pathogenesis of Facioscapulohumeral dystrophy. PLoS Genet. (2013)Jun;9(6):e1003550. | PMID: 23785297 | doi: 10.1371/journal.pgen.1003550 |
